


Helping you differentiate between IQ, OQ and PQ and decide what is right for you.
Regulations require that quality is ensured throughout the life cycle of a product. This means that manufacturers are responsible for product quality from manufacture to until it is used by the patient. This requirement is standardised under EU regulations as good distribution practices (GDPs) and is best practice for the global movement of pharmaceutical materials and products. To ensure your equipment is fit for purpose, Regulators will look to see that an appropriate qualification has been performed.
Qualification and Validation are terms often used interchangeably but simply put; Qualification is concerned with Equipment/Components and Validation is concerned with Processes.
Qualification:
The act of proving and documenting that equipment or ancillary systems are properly installed, work correctly, and comply with specified requirements. Qualification is part of validation, but the individual qualification steps alone do not constitute process validation.
Validation:
A documented objective evidence that provides a high degree of assurance that a specific process will consistently produce a product meeting its predetermined specifications Manufacturing Process is made up of several distinct components; personnel, equipment, and systems.
By qualifying the components, one is validating the process. Therefore, Qualification is directly related to equipment, systems, or software, and Validation is directly related to the process.
Design Qualification (DQ): Documentary evidence that equipment is designed to be capable of GMP compliance
Installation Qualification (IQ): An evaluation that the equipment was installed correctly and meets the pre-defined criteria set down in the URS (User Requirements Specification)
Operational Qualification (OQ): Verification that the equipment meets its operational requirements as described in Acceptance Criteria described in the protocol
Performance Qualification (PQ): Verification that the equipment performs when challenged under real-life operating conditions, usually with product in situ
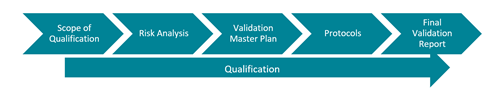
A robust Validation utilises a risk based strategy. Consider what the implications would be of, for example, temperature deviations in a controlled storage environment. Define the scope of the Qualification, in line with the VMP. Protocols will be prepared which will clearly define the Acceptance Criteria and the results will be summarised within the Final Report.
Thermal Mapping:
A part of the Operational Qualification and Performance Qualification process which is relevant to the following Industry Sectors:
- Pharmaceutical Production Areas
- Cold Chain Logistics including warehouses and vehicles
- Food Production
- Data centres
Thermal mapping typically measures temperature and RH parameters.

Guidelines note that to maintain regulatory compliance, any area used for the storage of products with a specified labelled storage requirement should be temperature mapped and a report generated to demonstrate the area is verified as fit for purpose.
Product safety and efficacy is a priority and regulatory authorities, and auditors will look for evidence of an understanding of the need to perform temperature mapping evaluation of areas used to store temperature-sensitive goods.
- Maintain Compliance with Regulations
- Identify hot/cold spots and potential breaches
- Trend data year on year to identify patterns
- In conjunction with permanent monitoring, use mapping data to identify equipment failures
All environmental controlled areas used to store time and temperature-sensitive products require Qualification:
- Stability Storage Chambers
- Stability Cabinets
- Fridges
- Freezers
- Cold Rooms
- Incubators
If you would like to learn more about how we can help with your Validation requirements contact us or call +44 (0) 1706 716710.
References:
- EU Guidelines of 5 November 2013 on Good Distribution Practice of Medicinal Products for Human Use (2013/C 343/01) Guidelines of 5 November 2013 on Good Distribution Practice of medicinal products for human useText with EEA relevance (europa.eu)
- WHO guidelines for Temperature Mapping of Storage Areas supplement_8.pdf (who.int)
- HPRA guidelines for Control and Monitoring of Storage and Transportation Temperature Conditions for Medicinal Products and Active Substances
- Current Good Manufacturing Practice (CGMP) Regulations- Title 21 of Code of Federal Regulations
- 21 CFR Part 210. Current Good Manufacturing Practice in Manufacturing Processing, packing, or Holding of Drugs.
- 21 CFR Part 211. Current Good Manufacturing Practice for Finished Pharmaceuticals.


